식품의약품안전처 고시 제2019 - 295호
「의료기기법」 제13조제1항, 제15조제6항, 제31조제1항 및 같은 법 시행규칙 제27조제1항, 제33조제1항, 제51조제4항에 따른 「의료기기 부작용 등 안전성 정보 관리에 관한 규정」(식품의약품안전처고시 제2019-231호, 2019.4.30)을 다음과 같이 개정 고시합니다.
2019년 6월 12일
식품의약품안전처장
의료기기 부작용 등 안전성 정보 관리에 관한 규정
의료기기 부작용 등 안전성 정보에 관한 규정 일부를 다음과 같이 개정한다.
제5조제2항 및 제3항을 각각 삭제하고, 같은 조 제4항, 제5항 및 제6항을 각각 제2항, 제3항 및 제4항으로 하며, 같은 조 제4항(종전의 제6항) 중 “제5항까지”를 “제3항까지”로 한다.
제5조의2를 다음과 같이 신설한다.
제5조의2(안내문 통지 등) ① 시행규칙 제27조제1항 및 제33조제1항에 따라 의료기기 제조업자 및 수입업자가 출고된 의료기기의 사용과 관련하여 의료기관(소비자)에게 주의사항 등을 알리려는 경우에는 별지 제1호의2 서식의 보고서에 해당 의료기기의 정보, 사용자가 취할 조치 등을 포함한 안내문을 첨부하여 식약처장에게 제출하여야 한다. 다만, 법 제31조에 따라 회수계획을 보고한 경우에는 이를 생략할 수 있다.
② 의료기기 제조업자 또는 수입업자는 안내문 통지를 완료한 경우 별지 제1호의2 서식의 보고서에 통지 결과 및 통지 사실을 증명할 수 있는 자료를 첨부하여 식약처장에게 제출하여야 한다.
③ 제1항 및 제2항에 따른 안내문 통지 보고는 우편·팩스·정보통신망 등의 방법으로 할 수 있다.
제6조를 삭제한다.
별지 제1호의2서식을 별지와 같이 신설한다.
별지 제2호 서식을 별지와 같이 한다.
부 칙
제1조(시행일) 이 고시는 고시한 날부터 시행한다.
제2조(경과조치) 이 고시 시행일 당시 종전의 규정에 따라 의료기기 이상사례 보고서를 접수한 경우에는 개정규정에도 불구하고 종전의 규정에 따른다.
■ 의료기기 부작용 등 안전성 정보 관리에 관한 규정 [별지 1호의2서식] 전자민원창구(emed.mfds.go.kr)에서도 신청할 수 있습니다.
의료기기 안내문 통지 보고서(의료기기 제조·수입업체)
※ 뒷쪽의 작성 시 참고사항을 확인하신 후 작성하시기 바랍니다.


|
보고 종류
|
□ 최초보고 ( 년 월 일)
□ 추가보고 ( 년 월 일) □ 최종보고 ( 년 월 일) |
||||
|
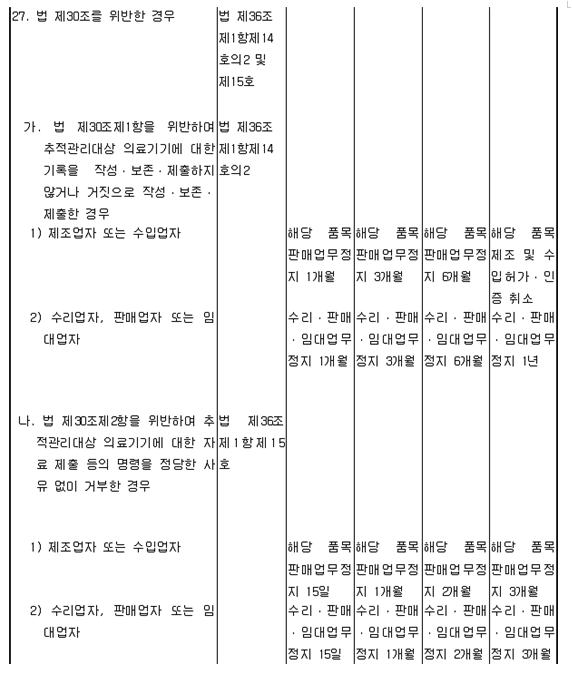
|
|||||
|
|
|||||
|
보고자 유형
|
의료기기취급자
|
||||
|
□ 의료기기제조업자 □ 의료기기수입업자
|
|||||
|
보
고 자 정 보 |
보고 업체명
|
|
성명
|
|
|
|
전화번호
|
|
E-mail
|
|
||
|
이상사례 보고 여부: □유 □무(사유: )
|
|||||
|
|
|||||
|
|
|||||
|
의
료 기 기 정 보 |
제 품 명
|
품 목 명
|
모 델 명
|
||
|
|
|
||||
|
분류번호
|
|
등 급
|
|
||
|
허가번호
|
|
제조번호
(Lot 번호) |
|
||
|
인체이식형 의료기기
|
□ 예 □ 아니오
|
||||
|
회사명/
제조원 (수입의 경우) |
|
||||
|
안
내 문 통 지 정 보 |
안내문 통지 결정일
|
년 월 일
|
|||
|
세부내용
|
|
||||
|
조치대상
|
의료기관(소비자) 수
|
|
|||
|
대상 제품 수
|
수입량
|
(단위)
|
|||
|
판매량
|
(단위)
|
||||
|
조치일정
|
안내문 통지 시작일
|
|
|||
|
안내문 통지 종료 예정일
|
|
||||
|
안내문 통지 종료일
|
|
||||
|
첨부자료
|
|
||||
210mm×297mm(백상지(80g/㎡) 또는 중질지(80g/㎡))
※ 작성 시 참고사항
1. 보고자의 개인정보는 식약처에 의해 엄격히 보호됩니다.
2. 보고 종류
2-1. 의료기기 사용과 관련하여 위해 방지를 목적으로 의료기기 취급자 및 사용자에게 주의사항 등을 알려야 하는 경우, 사용자가 취할 조치 등을 포함한 안내문 통지 계획을 최초보고 합니다.
2-2. 안내문 통지를 완료한 경우 통지 결과 및 통지 사실을 증명할 수 있는 자료를 최종보고 시 제출합니다.
2-3. 안내문의 내용, 조치 대상, 조치 일정의 수정이 있을 경우 추가보고를 실시하거나 최종보고에 반영하여 보고합니다.
2-4. 안내문 통지 최초보고 시 이미 안내문 통지가 종료된 경우 최종보고를 선택하여 작성할 수 있습니다.
3. 보고자 정보
3-1. 최근 1년 이내 의료기기로 인한 이상사례가 발생하여 식약처에 관련 보고한 경우 이상사례 보고여부에‘유’를 선택합니다.
3-2. 이상사례가 발생과 무관하지만 의료기기 취급자 및 사용자에게 주의사항 등을 알려야 하는 경우 이상사례 보고여부에‘무’를 선택하고 해당 사유를 작성합니다.
4. 안내문 통지 정보
4-1. 안내문 통지 결정일은 이상사례 발생 등에 따라 제조업자 혹은 수입업자의 경우 해외 제조원이 안내문 통지를 결정한 일자를 의미합니다.
4-2. 세부내용은 제조업자 또는 해외제조원에서 의료기기 취급자, 사용자 등을 대상으로 알리고자 하는 의료기기 안전사용을 위한 조치사항에 대한 세부내용(주의사항, 권고사항)을 상세하게 작성하여 제출합니다.
4-3. 안내문 통지가 필요한 의료기관(소비자), 대상 제품 수를 정확하게 파악하여 보고하되, 최초보고 시에는 추정치를 입력할 수 있습니다.
4-4. 안내문 통지 시작일은 의료기관 취급자, 사용자 등에 안내문 통지를 시작하는 일자를 의미하며, 추가보고를 통해 안내문 통지 종료예정일을 수정할 수 있습니다.
4-5. 안내문 통지 종료일은 최종보고 시에 작성하며, 의료기기 취급자 및 사용자에게 계획했던 안내문 통지가 모두 완료된 일자를 의미합니다.
5. 첨부 자료
5-1. 최초 및 추가보고 시에는 의료기관 취급자, 사용자 등에게 알리고자 하는 주의사항, 권고사항 등이 포함된 안내문(해외 제조원 원문은 번역본으로 제출할 것)을 첨부합니다.
5-2. 최종보고시에는 안내문 통지 증빙서류(업체 공문 또는 우편 발송 증명서 또는 소비자 서명이 포함된 확인서 등)를 첨부합니다.
6. 기입란이 부족한 경우에는 별지에 기입하여 주십시오.
■ 의료기기 부작용 등 안전성 정보 관리에 관한 규정 [별지 2호서식] 전자민원창구(emed.mfds.go.kr)에서도 신청할 수 있습니다.
의료기기 이상사례 신고서(소비자)
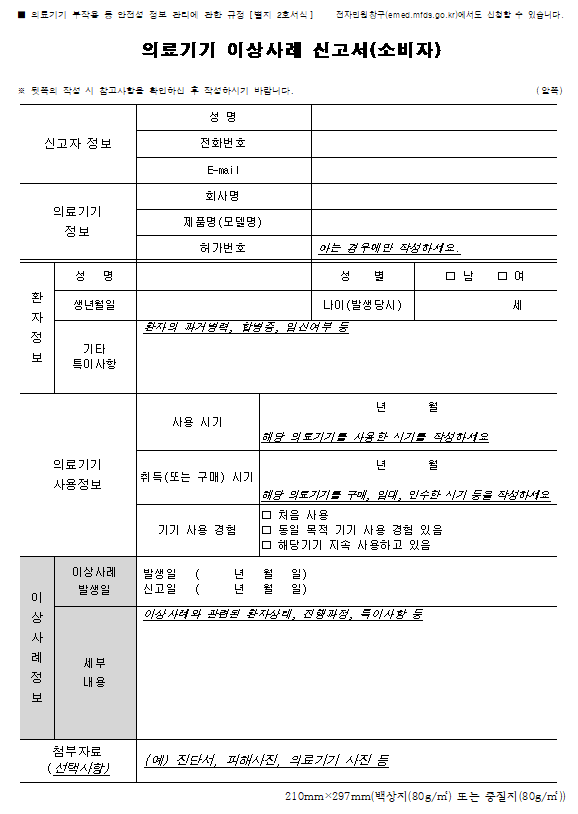
※ 뒷쪽의 작성 시 참고사항을 확인하신 후 작성하시기 바랍니다. (앞쪽)
|
신고자 정보
|
성 명
|
|
|||
|
전화번호
|
|
||||
|
E-mail
|
|
||||
|
의료기기
정보 |
회사명
|
|
|||
|
제품명(모델명)
|
|
||||
|
허가번호
|
아는 경우에만 작성하세요.
|
||||
|
환
자 정 보 |
성 명
|
|
성 별
|
□ 남 □ 여
|
|
|
생년월일
|
|
나이(발생당시)
|
세
|
||
|
기타
특이사항 |
환자의 과거병력, 합병증, 임신여부 등
|
||||
|
의료기기
사용정보 |
사용 시기
|
년 월
해당 의료기기를 사용한 시기를 작성하세요 |
|||
|
취득(또는 구매) 시기
|
년 월
해당 의료기기를 구매, 임대, 인수한 시기 등을 작성하세요 |
||||
|
기기 사용 경험
|
□ 처음 사용
□ 동일 목적 기기 사용 경험 있음 □ 해당기기 지속 사용하고 있음 |
||||
|
이
상 사 례 정 보 |
이상사례 발생일
|
발생일 ( 년 월 일)
신고일 ( 년 월 일) |
|||
|
세부
내용 |
이상사례와 관련된 환자상태, 진행과정, 특이사항 등
|
||||
|
첨부자료
(선택사항) |
(예) 진단서, 피해사진, 의료기기 사진 등
|
||||
210mm×297mm(백상지(80g/㎡) 또는 중질지(80g/㎡))
※ 작성 시 참고사항
1. 환자 및 신고자의 개인정보는 식약처에 의해 엄격히 보호됩니다.
2. 의료기기정보
소비자의 경우 회사명에 의료기기 판매업자를 기재할 수 있습니다.
3. 환자정보
3-1. 환자란 의료기기 사용과 관련하여 바람직하지 않은 건강영향 또는 임상영향을 받은 모든 사람을 의미합니다. (예: 의료인, 소비자 등 의료기기 사용자와 간병인, 보호자 포함)
3-2. 환자의 성명은 개인 식별이 불가능한 형태로 기입할 수 있습니다. (예: 홍길동→ ㅎㄱㄷ, HGD 등)
3-3. 환자정보 중 성별, 생년월일을 정확히 알 수 없는 경우 기입하지 않으셔도 되며, 나이를 정확히 알 수 없는 경우 연령대 기재가 가능합니다.
4. 이상사례 정보
4-1. 발생일은 이상사례가 발생한 일자를 말하여, 신고일은 신고자가 이상사례를 신고한 일자를 말합니다.
4-2. 세부내용은 이상사례가 발생하기까지의 경과와 환자상태를 의미합니다.
5. 첨부자료는 이상사례 정보와 의료기기 정보를 파악하는데 도움이 될 수 있는 진단서, 피해사진, 의료기기 사진 등을 의미합니다.
6. 불분명한 사항에 대해서는 기입하지 않으셔도 됩니다.
7. 기입란이 부족한 경우에는 별지에 기입하여 주십시오.
신ㆍ구조문대비표
|
현 행
|
개 정 안
|
|
|
|
|
제5조(이상사례의 보고) ① (생 략)
|
제5조(이상사례의 보고) ① (현행과 같음)
|
|
② 제1항에 따라 이상사례를 보고하려는 의료기기 제조업자 및 수입업자가 출고된 의료기기의 사용과 관련하여 위해 방지를 목적으로 의료기기 취급자 및 사용자에게 주의사항 등을 알려야 하는 경우에는 해당 의료기기 정보, 이상사례의 세부내용, 사용자가 취할 조치 등을 포함한 안내문을 추가로 첨부하여야 한다. 다만, 법 제31조에 따라 회수계획을 보고한 경우에는 이를 생략할 수 있다.
|
<삭 제>
|
|
③ 제2항에 따라 의료기기 제조업자 또는 수입업자가 의료기기취급자 및 사용자에게 안내문 통지를 완료한 경우 통지 결과 및 통지 사실을 증명할 수 있는 자료를 식약처장에게 제출하여야 한다.
|
<삭 제>
|
|
④ㆍ⑤ (생 략)
|
②ㆍ③ (현행 제4항 및 제5항과 같음)
|
|
⑥ 제1항부터 제5항까지에 따른 보고는 우편·팩스·정보통신망 등의 방법으로 할 수 있다.
|
④ ------ 제3항까지---------------------------------------------------.
|
|
<신 설>
|
제5조의2(안내문 통지 등) ① 시행규칙 제27조제1항 및 제33조제1항에 따라 의료기기 제조업자 및 수입업자가 출고된 의료기기의 사용과 관련하여 의료기관(소비자)에게 주의사항 등을 알리려는 경우에는 별지 제1호의2 서식의 보고서에 해당 의료기기의 정보, 사용자가 취할 조치 등을 포함한 안내문을 첨부하여 식약처장에게 제출하여야 한다. 다만, 법 제31조에 따라 회수계획을 보고한 경우에는 이를 생략할 수 있다.
② 의료기기 제조업자 또는 수입업자는 안내문 통지를 완료한 경우 별지 제1호의2 서식의 보고서에 통지 결과 및 통지 사실을 증명할 수 있는 자료를 첨부하여 식약처장에게 제출하여야 한다. ③ 제1항 및 제2항에 따른 안내문 통지 보고는 우편·팩스·정보통신망 등의 방법으로 할 수 있다. |
|
제6조(자료의 보완) ① 식약처장은 제5조에 따른 부작용 등 안전성 정보의 보고가 이 규정에 적합하지 아니하거나 추가 자료가 필요하다고 판단되는 경우 보완기간을 30일 이내로 하여 자료의 보완을 요구할 수 있다.
② 제1항에도 불구하고 보완요구를 받은 자가 원인조사, 해외 제조원의 자료 확보 등의 이유로 보완에 필요한 기간을 명시하여 기간 연장을 요청하는 경우에는 이를 고려하여 다시 보완기간을 정하여야 한다. 이 경우 민원인의 기간 연장 요청은 2회로 한정하며, 다시 정한 보완기간 내에 보완하지 아니한 경우에는 10일 이내에 다시 보완하도록 요구할 수 있다. |
<삭 제>
|
|
|
|
'의료기기 뉴스' 카테고리의 다른 글
| 창상피복재의 품목별 건강보험 분류 안내서 (2019.07) (0) | 2022.03.11 |
|---|---|
| 체외진단용 의료기기 변경 허가 관련 민원인 안내서 개정 (2019. 6. 28 ) (0) | 2022.03.11 |
| 의료기기통합정보 등록 시스템 사용자 교육 자료 (0) | 2022.02.27 |
| 「의료기기법 시행규칙」 개정 [시행 2019. 6. 12] [총리령 제1542호, 2019. 6. 12, 일부개정] (0) | 2022.02.27 |
| 「의료기기 통합정보 관리 등에 관한 규정」 [2019-46호 2019.06.12 ] (0) | 2022.02.22 |